
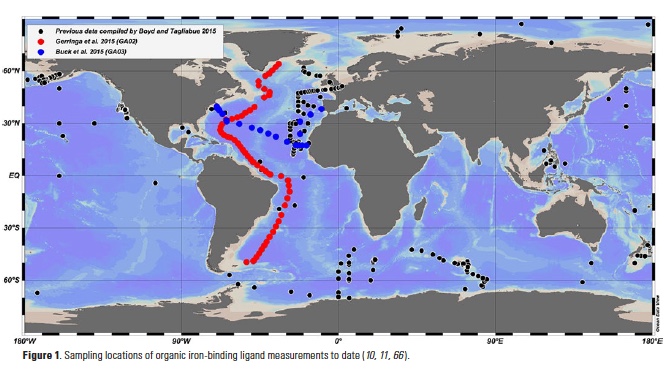
Iron is a limiting nutrient for phytoplankton in nearly half of the global surface ocean, and much attention has been paid to the biogeochemical cycling of iron in seawater since suitable trace metal-clean sampling and analysis procedures were developed (1). The organic complexation of dissolved iron, in particular, has emerged as an inherent feature of iron chemistry in the oceans (2-5), and iron speciation measurements are increasingly incorporated into field studies (Fig. 1). The integration of organic iron-binding ligands into biogeochemical models improves their ability to reproduce global dissolved iron distributions (6), and changes in ligand concentrations in the Southern Ocean can have a more pronounced impact on atmospheric CO2 in model studies than changes in iron supply terms from hydrothermal and dust sources (7). Indeed, field distribution measurements, targeted experimental studies and modeling efforts over the last twenty years have left little doubt that organic ligands are a critical factor in the global biogeochemical cycling of iron. Here we highlight some of the features of iron-binding ligand distributions in the Atlantic from the unprecedented basin-scale datasets coming out of the GEOTRACES program and attempt to elucidate some of the sources and sinks of iron-binding organic ligands in the oceans.
Dissolved iron speciation
The speciation of dissolved iron (Fe), which describes the chemical forms or species of iron in a filtered (typically <0.2 μm) sample, includes both inorganic (Fe´) and organic (FeL) components. In the oceans, iron speciation studies consistently report that nearly all dissolved iron (>99.9%) is organically complexed (8). This is perhaps not surprising given that the inorganic speciation of iron in oxygenated seawater is dominated by hydrolysis reactions leading to iron precipitation and very low inorganic iron solubility (~0.1 nmol L-1) under most ocean conditions (9).
Studies of organic iron speciation in seawater use an electrochemical technique, competitive ligand exchange-adsorptive cathodic stripping voltammetry (CLE-ACSV), to determine the concentrations and conditional stability constants of iron-binding organic ligands. This is accomplished by titrating iron-binding ligands in a sample with additions of iron and competing against any natural iron-ligand complexes with an added well-characterized ‘competitive’ ligand, which forms an electroactive complex with iron that can be measured at the surface of a hanging mercury drop electrode. Titrations often depict no measurable iron bound to the competitive ligand for the first few iron additions, reflecting the presence of excess strong iron-binding ligands in most seawater samples (8). These ligands are described as ligand classes, L1, L2, L3, L4, defined by the conditional stability constants determined by CLE-ACSV, with L1 and L2-type ligands the strongest iron-binding organic ligands
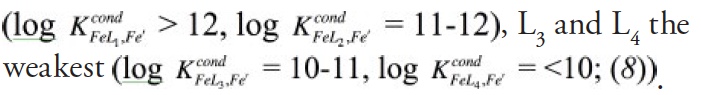
Iron speciation in the Atlantic: Observations from recent GEOTRACES efforts
The iron speciation datasets emerging from the GEOTRACES program allow a first look at basin-scale distributions of iron-binding ligands in the oceans. Iron speciation datasets from GEOTRACES Sections GA02 (10) and GA03 (11) from the Dutch and U.S. GEOTRACES programs, respectively, document the ubiquitous nature of iron-binding ligands in the Atlantic basin. In particular, both datasets evince the presence of strong, L1-type ligands throughout the water column and no discernible trend with depth in the conditional stability constants for these ligands (10, 11). These observations support the emerging picture from many other field studies of a strong iron-binding ligand pool that is not necessarily restricted to the surface ocean or euphotic zone (Fig. 2) (8).
Elevated dissolved iron and aluminum concentrations in surface waters across the GA03 zonal section, particularly near the center of the basin, demonstrate the widespread contribution of dust deposition to the iron inventory in the Atlantic (12). Iron isotope studies indicate that 71-87% of the dissolved iron along the entire GEOTRACES GA03 section was attributable to dust (13). Water-soluble organic matter characterized from the surfaces of aerosols collected on GA03 exhibited structural differences between aerosol sources that were consistent with their iron solubilities (14), and organic complexation of some of the leached iron was observed in seawater leaches of these aerosols (15). Recent studies using model ligands highlight the particular importance of stronger iron-binding ligands in the stabilization of iron leached from natural aerosols (16).
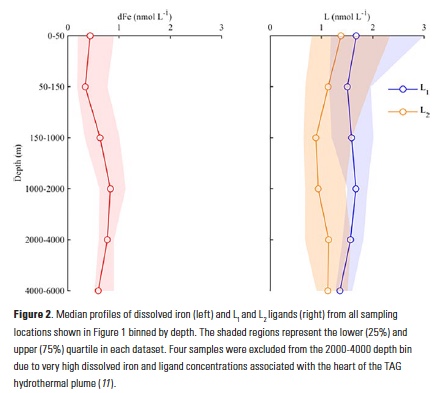
In the Atlantic GEOTRACES sections, the highest concentrations of ligands in excess of dissolved iron ([L]-[Fe], or L´) were often measured at the surface, where dissolved iron concentrations were low (10, 11). The overall complexation capacity for iron, which is a function of both ligand concentration and the conditional stability constant, also tended to be high in the upper water column. Antarctic Intermediate Water (AAIW) stands out in both datasets as exhibiting higher complexation capacity for dissolved iron than the surrounding water masses (10, 11). These waters originate from highly productive surface waters, and elevated ligand concentrations subducted with these water masses may be the result of higher strong iron-binding ligand concentrations commonly observed in and around chlorophyll maxima and in incubation experiments of iron-stressed diatom communities (see (8) and references therein). In a compilation of three iron speciation datasets that extend from the Arctic (17) down through the Western Atlantic (10) and into the Antarctic (18), higher ligand concentrations were reported at high latitudes relative to low latitudes, with the strongest (highest conditional stability constants) excess ligands measured in the Antarctic (10), and larger excesses of weaker ligands in the Arctic (10, 17).
Excess ligand concentrations in the Atlantic usually decreased with depth as dissolved iron concentrations increased, consistent with saturation of excess ligands with iron (10, 11). A north-south trend of decreasing ligands and excess ligands was reported in the GA02 Western Atlantic meridional section, which was clearly depicted in the samples collected from the North Atlantic Deep Water (NADW) along the section (10). In the GA03 zonal section of the North Atlantic (11), excess ligand concentrations in the water column were on the high end of the two datasets, consistent with the northern end of the GA02 meridional section (10). If anything, organic matter remineralization appeared to be a source of weaker L3-type iron-binding ligands in the GA03 zonal section dataset (11).
Excess ligand concentrations in the North Atlantic exhibited local minima in the heart of the oxygen minimum zone west of Mauritania (11), possibly due to scavenging of ligand complexes on sinking particles (19), or elevated reduced iron(II) concentrations complexing the excess ligands (20). It is unclear how much of the iron-binding ligand pool measured by CLE-ACSV may also bind iron(II), or what chemical form of iron(II) is present in these samples, though some may be biogenic (21). One of the most pronounced features in the iron(II) data from GA03 is the exceedingly high iron(II) concentrations in the TAG hydrothermal plume samples, where elevated dissolved iron was ~80% colloidal-sized (0.02-0.2 μm size fraction) iron(II) species (20, 22). These iron(II) colloids are likely pyrite nanoparticles (23), which themselves may be stabilized by organic matter (24).
In the TAG plume samples collected along GA03, excess ligands were at a minimum in the highest iron samples of the plume, but the conditional stability constants of the excess ligands that were detected in these samples were among the highest in the dataset, leading to an elevated complexation capacity for iron around the vent. It is likely that some of the elevated dissolved iron in these samples was not exchangeable with the added competitive ligand during the voltammetry measurements, which would lead to an overestimation of ligand parameters (8). It is also possible that some of this observed increase in complexation capacity in the plume reflects a microbial response to the iron-enriched plume (25). Several studies have now reported varying degrees of organic complexation of iron in hydrothermal vent plumes (10, 11, 26, 27), and while the cycling of ligands in these systems remains unclear, organic stabilization must be a key factor in determining the chemical speciation and transport of iron from these vents into the deep sea (28-30).
The increasingly rich database of iron-binding ligand distributions from individual field studies and the GEOTRACES program show the widespread organic complexation of iron in the oceans. These datasets also display the inherent complexity of ligand cycling, since ligands are at the interface between the dynamic biogeochemical cycles of both trace metals and organic matter in seawater.
Identity of iron-binding ligands
Electrochemistry (CLE-ACSV) measures ligand concentrations and conditional stability constants from a combination of titration and competition, and is the basis of most of our insights to date into the sources, sinks, and cycling of organic iron-binding ligands in the oceans. Mass spectrometry-based techniques, on the other hand, are increasingly being employed to identify the chemical structures of natural iron-ligand complexes in the oceans. Results from both of these approaches appear to be converging on similar descriptions of the iron-binding ligand pool, one which comprises a mixture of defined biomolecules with high affinities for iron (e.g., siderophores, heme) and weaker iron-binding, ill-defined compounds with high chemical heterogeneity (e.g., humic substances, polysaccharides) (Fig. 3; (8)).
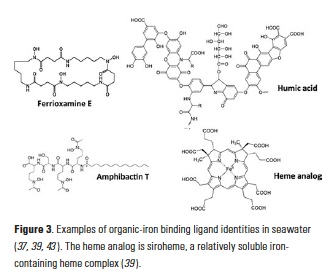
Siderophores are small iron-binding ligands widely produced by bacteria to acquire iron from the environment (31), including the marine environment (32, 33). Model siderophores are typically, though not exclusively, characterized as among the strongest L1-type ligands measured in CLE-ACSV (3, 8). Iron complexed by these discrete biomolecules can usually be chromatographically resolved, although isolation of these compounds from seawater is notoriously difficult. Ferrioxamines and amphibactins have been the most widely reported from the water column (Figure 3; (34-36)) and shipboard incubations (37), though marine bacteria cultures have produced a broader suite (32). The limited diversity and only picomolar concentrations of siderophores extracted from seawater compared to marine bacteria cultures likely reflects limitations in sampling, the extraction procedures available (8, 32), and detection of certain siderophore functional groups that are preferentially photodegraded in surface waters (38). Importantly, siderophores have been observed to persist throughout the water column below the euphotic zone (36) and are not restricted to low-iron waters (34). Additional discrete biomolecules, like heme or intracellular iron storage proteins, are also expected to contribute to the strong iron-binding ligand pool in seawater, though they may be prone to particle adsorption and aggregation processes, making them more likely to be found in the colloidal and particulate phases (8, 39). Because of their role as intracellular iron-binding ligands, these molecules are Science usually released to the extracellular environment as iron complexes, rather than as free ligands. Similarly, viruses may even constitute a component of colloidal organically complexed iron (40).
Unlike siderophores, humic substances and polysaccharides are complex molecules with high heterogeneity and complexity (Fig. 3), which cannot generally be resolved chromatographically, but represent a large component of the natural organic matter pool (41). Some of these molecules actually form electroactive complexes with iron and have been directly measured by electrochemistry in estuarine, coastal, and deep open ocean waters (42-44). Suwannee River Fulvic Acid (SRFA), a suspected component of the refractory dissolved organic matter pool (45), has been identified as a model ligand that can be used to reproduce the peak of natural electroactive iron complexes (46); exopolymeric substances can similarly form electroactive iron complexes (47). The conditional stability constants for exopolysaccharides and SRFA determined by CLE-ACSV generally fall under the L2 to L4 ligand class definitions (8).
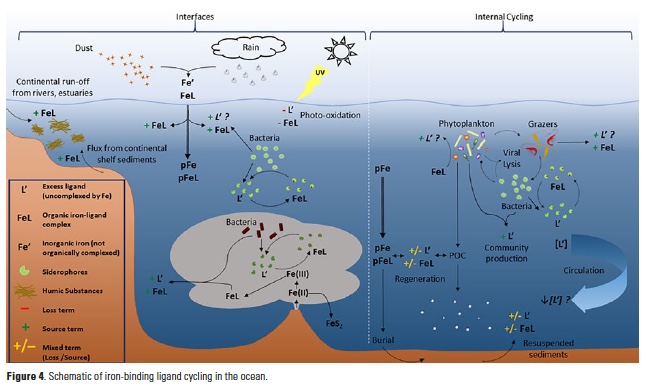
Ligand processes at ocean interfaces (Fig. 4)
Photochemical degradation of natural iron-binding ligands is variable in field studies (48, 49), and may account for sea surface minima in ligand concentrations observed in some profiles (8). Experimental studies indicate that siderophore photolability depends on chemical structure and whether the siderophore is bound to iron (FeL) or not (L´) (38). Humic substances, on the other hand, are universally photoreactive by the nature of their molecular structure (50), though the iron-binding ability of their photoproducts is unknown. Dust deposition and rainfall may serve as ligand sources if depositing strong iron-binding ligands in addition to their iron loads (14, 51-54), or stimulating ligand production by surface microbial communities (55, 56), which may be critical for stabilizing atmospherically-derived iron in surface waters (16). Inorganic iron additions in mesoscale fertilization experiments have also been shown to stimulate ligand production in the fertilized waters (8, 57). Similarly, a microbial iron cycle fueled by hydrothermal iron inputs at the crust-ocean interface has recently been suggested (25) to support the organic stabilization and transport of dissolved iron in plumes extending remarkable distances from vent systems (28, 30).
Along the coastal margins, organically complexed iron is delivered to the coastal ocean from river plumes, estuaries, and shelf sediments, often along with excess weaker iron-binding ligands, including humic substances (8, 42, 44, 58). Elevated excess iron-binding ligands were observed in bottom waters of several of the GA03 stations in the North Atlantic (11) that were also local maxima in excess copper-binding ligands (59), indicating overlap in the ligand pool between these two bioactive elements (44).
Internal ligand cycling (Fig. 4)
The low solubility of inorganic iron, and the overwhelming organic complexation of the dissolved iron pool by a diverse suite of ligands has significant implications for iron bioavailability to marine phytoplankton (60, 61). The high biological demand for iron by phytoplankton and heterotrophic bacteria in turn supports a myriad of iron acquisition strategies, which are largely mediated by organic complexation (33, 61). High excess ligand concentrations in low-iron waters may potentially result from ligand production, iron uptake from a ligand complex, or both. Production of excess iron-binding ligands has been observed under a range of iron conditions, with iron additions in large-scale iron fertilization experiments (57), and in iron-stressed diatom communities (49, 62, 63), perhaps indicative of a community iron cycle including diatom-associated bacterial communities (64).
Grazing, viral lysis, and organic matter remineralization are likely important sources of weaker iron-binding ligands (e.g., humic-like substances, exopolysaccharides, or intracellular organic iron complexes like heme) to the ocean interior (8, 19). Mounting evidence points to bacterial production of the strongest ligands observed in seawater as an iron uptake strategy (8, 49, 57). Organic matter remineralization may similarly be a source of strong iron-binding ligands to the entire water column, given that siderophore production by heterotrophic bacteria is not necessarily restricted to the surface ocean and may be associated with ‘hot spots’ of sinking organic matter in the deep sea (65).
The extent to which iron-binding ligands are remineralized themselves is unknown. Excess ligand concentrations tended to decrease with depth in the Western North Atlantic as dissolved iron concentrations increased (10, 11). Decreasing total ligand concentrations in NADW samples were negatively correlated, albeit weakly, with apparent oxygen utilization (AOU) along the GA02 meridional section (10), implying some ligand remineralization during circulation since dilution was not expected to impact ligand concentrations. Gerringa et al. (2015) calculated a residence time on the order of 103 years for iron-binding ligands in the NADW, up to four times longer than that of dissolved iron, suggesting that particles must scavenge iron from strong organic complexes in the deep sea (10). Overall, the interactions between iron-binding ligands and sinking particles, whether lithogenic or biogenic in origin, are largely uncharacterized. These particles likely serve as both sources and sinks of iron-binding ligands (19) depending on the nature of the ligands, particles and biological communities involved.
Conclusions
Detailed large-scale datasets from the Atlantic Ocean have given us an unparalleled view of ligand cycling in this basin. These studies have enabled us to take a holistic look at ligand sources and sinks and internal cycling for the first time, and new paradigms have emerged. Biological contributions to the ligand pool are clear across nearly all ligand datasets. Although the direct connection between the organisms responsible for ligand production and the compounds they produce is still uncertain, marine microorganisms appear to be active producers of strong iron-binding ligands that influence iron cycling through the water column. Expansion of basin-scale datasets to the other basins and collaborative experimental studies to elucidate the mechanisms of ligand cycling behind the basin-scale distributions, some of which have been described here for the Atlantic, will improve under standing of the cycling of organic iron-binding ligands and inform global biogeochemical models of iron and carbon cycles.
Authors
Kristen N. Buck (Univ. of South Florida, College of Marine Science), Chelsea Bonnain (Univ. of South Florida, College of Marine Science), and Randelle M. Bundy (Woods Hole Oceanographic Institution)
Acknowledgments
The authors thank Kathy Barbeau for her helpful comments on the text. This work was funded in part by the National Science Foundation through an award to KNB, OCE-0927453.
References
- K. W. Bruland et al., Analytica Chimica Acta 105, 233-245 (1979).
- M. Gledhill, C. M. G. van den Berg, Marine Chemistry 47, 41-54 (1994).
- E. L. Rue, K. W. Bruland, Marine Chemistry 50, 117-138 (1995).
- C. M. G. van den Berg, Marine Chemistry 50, 139-157 (1995).
- J. Wu, G. W. Luther III, Marine Chemistry 50, 159-177 (1995).
- A. Tagliabue et al., Global Biogeochemical Cycles 30, 149-174 (2016).
- A. Tagliabue et al., Geophysical Research Letters 41, 920-926 (2014).
- M. Gledhill, K. N. Buck, Frontiers in Microbiology: Microbiological Chemistry 3, Article 69 (2012).
- X. Liu, F. J. Millero, Marine Chemistry 77, 43-54 (2002).
- L. J. A. Gerringa et al., Marine Chemistry 177, 434-446 (2015).
- K. N. Buck et al., Deep-Sea Research II 116, 152-165 (2015).
- C. Measures et al., Deep-Sea Research II 116, 176-186 (2015).
- T. M. Conway, S. G. John, Nature 511, 212-215 (2014).
- A. S. Wozniak et al., Marine Chemistry 154, 24-33 (2013).
- K. N. Buck, A. M. Aguilar-Islas, (unpublished).
- M. P. Fishwick et al., Global Biogeochemical Cycles 28, 1235-1250 (2014).
- C.-E. Thuróczy et al., Journal of Geophysical Research 116, C10009 (2011).
- C.-E. Thuróczy et al., Deep-Sea Research II 58, 2695-2706 (2011).
- P. W. Boyd et al., Limnology and Oceanography 55, 1271-1288 (2010).
- P. N. Sedwick et al., Deep-Sea Research II 116, 166-175 (2015).
- B. P. von der Heyden et al., Environmental Science & Technology Letters 1, 387-392 (2014).
- J. N. Fitzsimmons et al., Deep-Sea Research II 116, 130-151 (2015).
- M. Yücel et al., Nature Geoscience 4, 367-371 (2011).
- B. M. Toner et al., Nature Geoscience 2, 197-201 (2009).
- M. Li et al., Nature Communications 5, 3192-3199 (2013).
- S. A. Bennett et al., Earth and Planetary Science Letters 270, 157- 167 (2008).
- J. A. Hawkes et al., Earth and Planetary Science Letters 375, 280- 290 (2013).
- J. N. Fitzsimmons et al., Proceedings of the National Academy of Sciences 111, 16654-16661 (2014).
- S. G. Sander, A. Koschinsky, Nature Geoscience 4, 145-150 (2011). Science
- J. A. Resing et al., Nature 523, 200-203 (2015).
- J. B. Neilands, Journal of Biological Chemistry 270, 26723-26726 (1995).
- J. M. Vraspir, A. Butler, Annual Review of Marine Science 1, 43-63 (2009).
- M. Sandy, A. Butler, Chemical Reviews 109, 4580-4595 (2009).
- E. Mawji et al., Environmental Science & Technology 42, 8675- 8680 (2008).
- R. M. Boiteau, Massachusetts Institute of Technology-Woods Hole Oceanography Institution, (2016).
- R. M. Bundy et al., Limnology and Oceanography, (in prep).
- M. Gledhill et al., Marine Chemistry 88, 75-83 (2004).
- K. Barbeau et al., Limnology and Oceanography 48, 1069-1078 (2003).
- S. L. Hogle et al., Metallomics 6, 1107-1120 (2014).
- C. Bonnain et al., Frontiers in Marine Science 3, article 82 (2016).
- L. I. Aluwihare et al., Nature 387, 166-169 (1997).
- R. M. Bundy et al., Marine Chemistry 173, 183-194 (2015).
- L. M. Laglera, C. M. G. Van den Berg, Limnology and Oceanography 54, 610-619 (2009).
- M. M. Abualhaija et al., Marine Chemistry 172, 46-56 (2015).
- N. Arakawa, L. Aluwihare, Environmental Science & Technology 49, 4097-4105 (2015).
- L. M. Laglera et al., Analytica Chimica Acta 599, 58-66 (2007).
- C. S. Hassler et al., Proceedings of the National Academy of Sciences 108, 1076-1081 (2011).
- K. Barbeau, Photochemistry and Photobiology 82, 1505-1516 (2006).
- R. M. Bundy et al., Frontiers in Marine Science 3, Article 27 (2016).
- P. G. Coble, Chemical Reviews 107, 402-418 (2007).
- A. C. Saydam, H. Z. Senyuva, Geophysical Research Letters 29, 1524 (2002).
- M. Cheize et al., Analytica Chimica Acta 736, 45-54 (2012).
- J. N. Fitzsimmons et al., Marine Chemistry, (2015 in press).
- J. D. Willey et al., Limnology and Oceanography 53, 1678-1684 (2008).
- T. Wagener et al., Geophysical Research Letters 35, L16601 (2008).
- L. J. A. Gerringa et al., Marine Chemistry 102, 276-290 (2006).
- C. L. Adly et al., Limnology and Oceanography 60, 136-148 (2015).
- M. E. Jones et al., Limnology and Oceanography 56, 1811-1823 (2011).
- J. E. Jacquot, J. W. Moffett, Deep-Sea Research II 116, 187-207 (2015).
- F. M. M. Morel et al., Limnology and Oceanography 53, 400-404 (2008).
- Y. Shaked, H. Lis, Frontiers in Microbiology 3, Article 123 (2012).
- K. N. Buck et al., Marine Chemistry 122, 148-159 (2010).
- A. L. King et al., Marine Chemistry 128-129, 1-12 (2012).
- S. A. Amin et al., Microbiology and Molecular Biology Reviews 76, 667-684 (2012).
- F. Azam, F. Malfatti, Nature Reviews 5, 782-791 (2007).
- P. W. Boyd, A. Tagliabue, Marine Chemistry 173, 52-66 (2015).

